Proteins are fundamental components of life’s molecular machinery, driving essential biological processes such as catalyzing chemical reactions, coordinating immune responses, and regulating gene expression. Their diverse functions power applications ranging from gene therapies and vaccines to industrial enzymes that support sectors like pharmaceuticals and agriculture. Each protein is encoded as a sequence of amino acids — a 20-letter alphabet that determines both its three-dimensional structure and its function. Protein engineers design or modify these sequences to create proteins with desired properties.
At Profluent, we build Protein Language Models (PLMs) that offer a data-driven framework for modeling the relationships between protein sequence, structure, and function. Our PLMs are trained on billions of proteins and trillions of amino acids from the Profluent Protein Atlas (PPA), the most extensive and carefully curated protein dataset available to date. Through large-scale training, PLMs implicitly learn evolutionary patterns shaped by natural selection over billions of years.
Profluent-E1 is our new family of retrieval-augmented PLMs (RA-PLMs) that enhances the PLM training paradigm by explicitly providing relevant evolutionary context in the form of homologous protein sequences, both during training and inference. In contrast to ProGen3, which is a generative model, E1 is an encoder model that learns representations of proteins useful for downstream tasks. E1 leverages the scale of the PPA and targeted architectural and training innovations to achieve state-of-the-art performance among encoder models trained exclusively on sequence data — both in single-sequence mode (without homologs at inference) and in retrieval-augmented mode.
We are releasing three E1 variants — with 150M, 300M, and 600M parameters — freely for both research and commercial use, enabling immediate application to tasks such as fitness prediction, structure prediction, and representation learning.
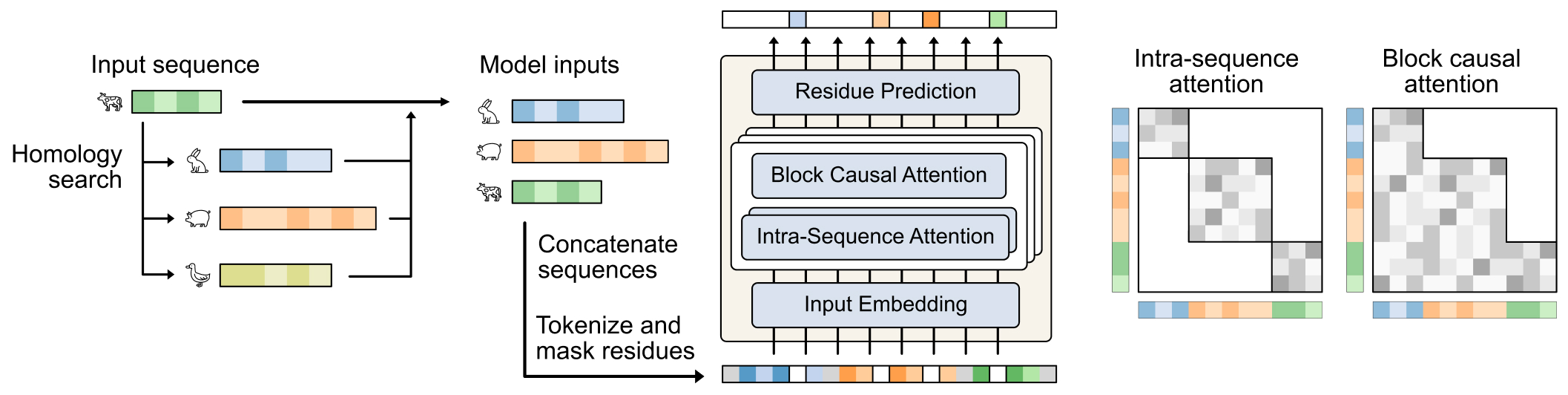
Setting a New Bar for Protein Encoder Models
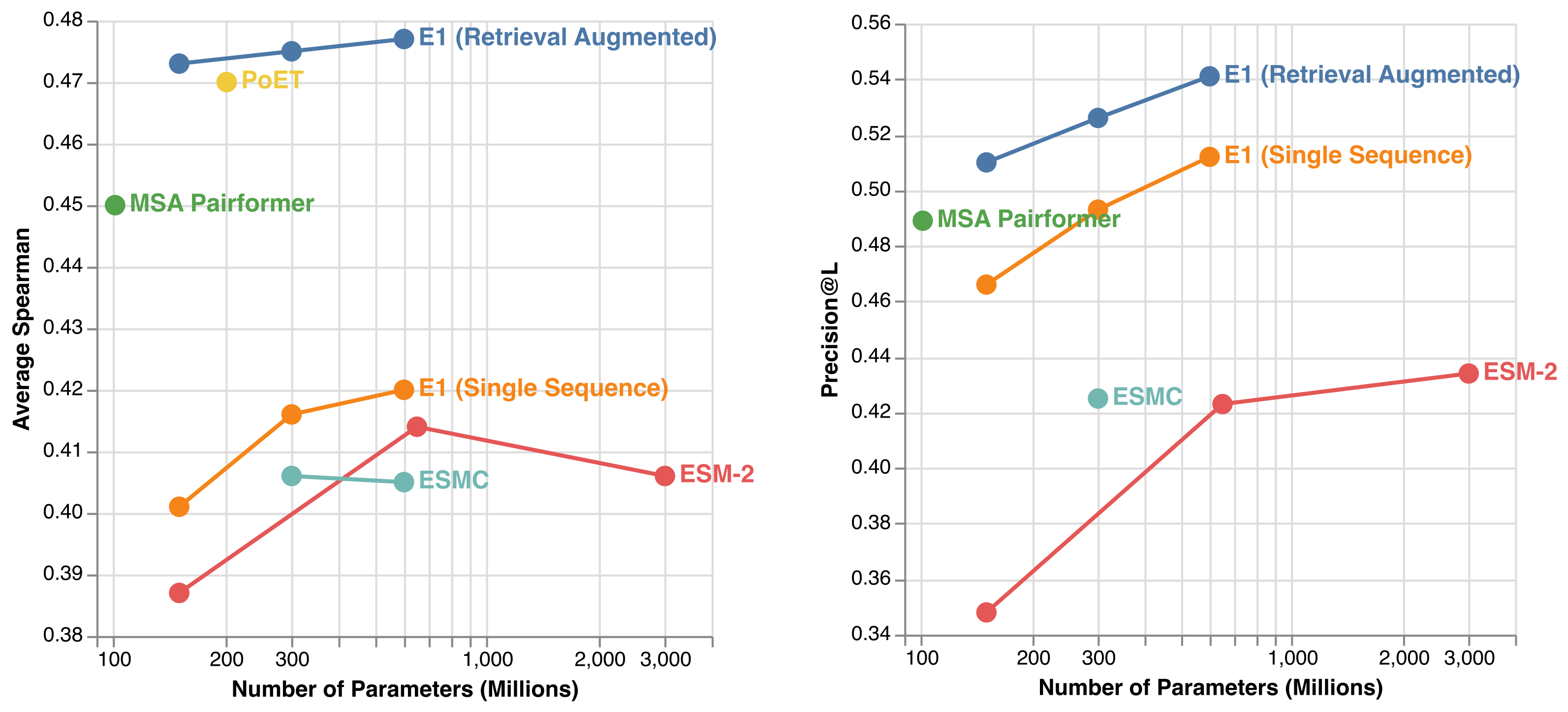
It has been shown that PLMs are effective zero-shot fitness predictors. In other words, they can accurately assess the impact of introducing specific mutations to a protein’s overall fitness (e.g. activity, expression, stability). To evaluate E1’s fitness prediction capabilities, we used the ProteinGym (v1.3) benchmark, which includes 217 experimental deep mutational scanning assays. At comparable model sizes, E1 outperforms all ESM-2 and ESM-C models in single-sequence mode — demonstrating that E1 can serve as a drop-in replacement for existing single-sequence encoder models (see left figure above). Retrieval augmentation further boosts E1’s accuracy, enabling it to surpass publicly available retrieval-augmented models such as MSA Pairformer and PoET.
We also assessed E1’s ability to capture protein structural information via long-range contact prediction, using sequences from CAMEO and CASP15 targets. In single-sequence mode, E1 outperforms the ESM family across model scales (see right figure above). Retrieval augmentation provides additional consistent gains, showing that E1 effectively leverages homologous sequences at inference time to better infer putative 3D contacts.
Taken together, the Profluent-E1 family demonstrates the continued value of research in improving protein language models. E1 provides a new foundational tool for AI-driven protein design, improving predictive performance and delivering practical utility across a broad range of protein engineering workflows.